
Najważniejsze informacje o serializacji leków
W celu zwiększenia nadzoru nad bezpieczeństwem leków Dyrektywa Fałszywkowa wprowadziła obowiązek umieszczania na większości leków na receptę dwóch rodzajów zabezpieczeń: unikalnego identyfikatora (UI) w postaci kodu dwuwymiarowego oraz elementów wskazujących na otwarcie opakowania (ATD).

Dyrektywa Parlamentu Europejskiego i Rady 2011/62/UE z dnia 8 czerwca 2011 r. zmieniająca dyrektywę 2001/83/WE w sprawie wspólnotowego kodeksu odnoszącego się do produktów leczniczych stosowanych u ludzi – w zakresie zapobiegania wprowadzaniu sfałszowanych produktów leczniczych do legalnego łańcucha dystrybucji nazywana Dyrektywą Fałszywkową lub FMD (Falsified Medicines Directive) została uchwalona w celu zabezpieczenia europejskiego łańcucha dystrybucyjnego przed wprowadzaniem sfałszowanych produktów leczniczych. Sfałszowanymi produktami leczniczymi mogą być leki zawierające mniejszą ilość substancji czynnych lub nie zawierające ich wcale, mogą zwierać inne substancje czynne. Sfałszowane mogą być również opakowania lub zabezpieczenia opakowań a także skradzione leki lub opakowania ponownie wprowadzone do obrotu.
Podstawowym źródłem sfałszowanych leków są sprzedawcy działający nielegalnie oferując je np. na bazarach lub w internecie. Mimo, to inspekcja farmaceutyczna a także inne organy wykrywają przypadki obecności sfałszowanych produktów w legalnym łańcuchu dystrybucji.
Jednym z celów Dyrektywy Fałszywkowej jest stworzenie europejskiego systemu baz danych poprzez który apteki, hurtownie oraz inne podmioty dostarczające leki pacjentom będą dokonywać weryfikacji autentyczności produktów medycznych przed ich wydaniem.
Obowiązek weryfikacji i wycofania kodów z bazy danych został nałożony na wszystkie osoby upoważnione lub uprawnione do wydawania pacjentom produktów leczniczych objętych serializacją. Dotyczy to zarówno hurtowników, aptekarzy w aptekach ogólnodostępnych i szpitalnych, działach farmacji, oraz podmioty wymienione w art. 23 Rozporządzenia Delegowanego 2016/161.
Na poziomie aptek mogą pojawić się koszty związane z dostosowaniem infrastruktury ich technicznej do wymogów aktów delegowanych, np. zakupem skanerów 2D lub dostosowaniem systemów informatycznych. Obowiązujące przepisy nie przewidują obowiązku poniesienia tych kosztów przed podmioty trzecie.
Naczelna Izba Aptekarska prowadzi intensywne prace mające na celu obniżenie kosztów ponoszonych przez apteki związanych z zakupem skanerów 2D oraz integracją sytemu baz danych z systemami egzystującymi w aptekach.
Wyznaczonym terminem obowiązywania nowych przepisów i rozpoczęcia prowadzenia weryfikacji autentyczności leków jest 9 lutego 2019 r.
Poniżej można zapoznać się z materiałem instruktażowym, obrazującym proces weryfikacji autentyczności leków serializowanych:
Poniżej znajdują się dokumenty regulujące wprowadzenie Dyrektywy Fałszywkowej:
Rozporządzenie delegowane Komisji UE
Dyrektywa Rarlamentu Europejskiego i Rady
Naczelna Izba Aptekarska informuje, iż Krajowa Organizacja Weryfikacji Autentyczności Leków (KOWAL) jest w trakcie bieżącego procesu nadawania certyfikatów dostępu do środowiska produkcyjnego PLMVS.
Z uwagi na dużą liczbę użytkowników końcowych (około 16 tys.) nadawanie certyfikatów jest procesem rozłożonym w czasie. Od grudnia 2018 r. certyfikaty są sukcesywnie nadawane i wysyłane.
Naczelna Izba Aptekarska przekazuje poniżej link do przygotowanego przez fundację KOWAL filmu instruktażowego, dotyczącego procesu generowania certyfikatów:
Proces pobrania certyfikatu do środowiska testowego (IQE) wygląda jak na zaprezentowanym powyżej filmie instruktażowym. Generowanie certyfikatu do środowiska produkcyjnego (PRD) odbywać się będzie dwuetapowo, zgodnie z informacją przekazaną tutaj.
Naczelna Izba Aptekarska ponadto przypomina, iż przed dniem 9 lutego 2019 r. każdy użytkownik końcowy musi być w posiadaniu oprogramowania umożliwiającego połączenie się z systemem PLMVS, pozwalającego na weryfikację i wycofanie serializowanych produktów leczniczych.
Najczęściej zadawane pytania
Komunikat dotyczący stosowania rozporządzenia Delegowanego Komisji (UE) 2016/161 z dnia 2 października 2015 r. uzupełniającego dyrektywę 2001/83/WE Parlamentu Europejskiego i Rady przez określenie szczegółowych zasad dotyczących zabezpieczeń umieszczanych na opakowaniach produktów leczniczych stosowanych u ludzi, (Dz.U.UE.L.2016.32.1 z dnia 2016.02.09).
W związku z bardzo licznymi pytaniami i monitami farmaceutów dotyczącymi wejścia w życie w dniu 9 lutego 2019 r. Rozporządzenia Delegowanego Komisji (UE) 2016/161 z dnia 2 października 2015 r. uzupełniającego dyrektywę 2001/83/WE Parlamentu Europejskiego i Rady przez określenie szczegółowych zasad dotyczących zabezpieczeń umieszczanych na opakowaniach produktów leczniczych stosowanych u ludzi (Dz.U.UE.L.2016.32.1 z dnia 2016.02.09), zwanego dalej „Rozporządzeniem”, Naczelna Izba Aptekarska zwraca uwagę na następujące okoliczności:
Przede wszystkim podkreślić należy, że Rozporządzenie ma zastosowanie do „produktów leczniczych, które zostały dopuszczone do sprzedaży lub dystrybucji” od 9 lutego 2019 r. Zdaniem Naczelnej Izby Aptekarskiej zwrot „dopuszczone do sprzedaży lub dystrybucji” oznacza czynność zwolnienia serii danego produktu leczniczego.
Zgodnie z § 48 Rozporządzenia, „produkty lecznicze, które zostały dopuszczone do sprzedaży lub dystrybucji” w państwie członkowskim bez zabezpieczeń przed datą rozpoczęcia stosowania niniejszego rozporządzenia w tym państwie członkowskim i nie zostały w późniejszym terminie ponownie zapakowane ani ponownie oznaczone, mogą być wprowadzane do obrotu, dystrybuowane i dostarczane pacjentom w tym państwie członkowskim aż do terminu ich ważności.
Zdaniem Naczelnej Izby Aptekarskiej, z powyższej normy wynika, że:
1) wszystkie „produkty lecznicze, które zostały dopuszczone do sprzedaży lub dystrybucji” przed 9 lutego 2019 r. i po tej dacie nie zostały ponownie zapakowane ani ponownie oznaczone, nie mają zabezpieczeń w rozumieniu Rozporządzenia, ponieważ takie zabezpieczenia mogły być wymagane najwcześniej w dniu 9 lutego 2019 r.;
2) w konsekwencji „produkty lecznicze, które zostały dopuszczone do sprzedaży lub dystrybucji” przed 9 lutego 2019 r. i po tej dacie nie zostały ponownie zapakowane ani ponownie oznaczone, nie podlegają weryfikacji autentyczności przewidzianej w Rozporządzeniu.
Każdy produkt leczniczy, co do którego apteka posiada informację, że spełnia wskazany powyżej warunek, tj. zwolnienie serii przed 9 lutym 2019 r. oraz brak po tej dacie ponownego zapakowania lub ponownego oznaczenia) może być spokojnie wydany pacjentowi bez weryfikacji autentyczności.
Zdaniem Naczelnej Izby Aptekarskiej bez wątpienia będą to leki znajdujące się na stanie magazynu apteki oraz leki, co do których hurtownia farmaceutyczna potwierdzi, że zwolnienie serii dokonane zostało przed 9 lutym 2019 r.
Jeżeli produkt leczniczy został „dopuszczony do sprzedaży lub dystrybucji” przed 9 lutego 2019 r., apteka ma prawo przyjąć, że lek ten nie posiada zabezpieczenia w rozumieniu Rozporządzenia, nie dotyczy go obowiązek weryfikacji zabezpieczeń i pozostaje w obrocie na podstawie art. 48 Rozporządzenia. Zgodnie z art. 546 § 1 Kodeksu cywilnego, sprzedawca leku obowiązany jest przed zawarciem umowy udzielić kupującemu, w tym aptece, potrzebnych wyjaśnień o stosunkach prawnych i faktycznych dotyczących rzeczy. Sprzedawca obowiązany jest wydać posiadane przez siebie dokumenty, które dotyczą rzeczy. Jeżeli treść takiego dokumentu dotyczy także innych rzeczy, sprzedawca obowiązany jest wydać uwierzytelniony wyciąg z dokumentu.
Co do produktów leczniczych, które zostały „dopuszczone do sprzedaży lub dystrybucji”, po dniu 9 lutego 2019 r. obowiązujące przepisy Rozporządzenia wymagają weryfikacji autentyczności.
Naczelna Izba Aptekarska posiada informacje, że poszczególne państwa członkowskie Unii Europejskiej wprowadzają na swoim terenie okres dostosowawczy do wymogów Rozporządzenia. Jedynym jednak podmiotem uprawnionym do podjęcia takich decyzji są stosowne organy władzy publicznej, tj. Minister Zdrowia oraz Główny Inspektor Farmaceutyczny.
Sprawa wprowadzenia okresu dostosowawczego do przepisów Rozporządzenia podnoszona była przez przedstawicieli Naczelnej Izby Aptekarskiej uczestniczących w spotkaniach Fundacji „KOWAL” dotyczących wdrożenia przepisów Rozporządzenia na obszarze Polski.
Na spotkaniach tych reprezentanci NIA jednoznacznie wypowiedzieli się za koniecznością wprowadzenia takiego okresu oraz potwierdzili, że dla farmaceutów jedynymi wiążącymi rozstrzygnięciami w tym zakresie są rozstrzygnięcia stosownych organów władzy.
Przedstawiając powyższe, Naczelna Izba Aptekarska informuje również, że na dzień dzisiejszy nie wprowadzone zostały żadne zmiany przepisów prawa w tym zakresie, w tym również dotyczące stosowania przez wojewódzkich inspektorów farmaceutycznych kar.
PRAWO NIE PRZEWIDUJE KAR DOTYCZĄCYCH BEZPOŚREDNIO NIEWYKONYWANIA OBOWIĄZKÓW OKREŚLONYCH W ROZPORZĄDZENIU.
Naczelna Izba Aptekarska będzie informować farmaceutów, na bieżąco o każdej decyzji podejmowanej w tym zakresie, a mającej wpływ na prawa i obowiązki farmaceutów.
Dyrektywa antyfałszywkowa w aptekach szpitalnych i w działach farmacji szpitalnej
Komunikat do Dyrektorów Szpitali posiadających aptekę szpitalną/dział farmacji szpitalnej w sprawie serializacji leków
Kluczowym środkiem w zakresie przeciwdziałania fałszowaniu produktów leczniczych w UE i ochrony legalnego łańcucha dystrybucji tych produktów jest kompleksowy system weryfikacji wprowadzony dyrektywą w sprawie sfałszowanych produktów leczniczych. Kompleksowa weryfikacja to system uwierzytelniania leków obejmujący obowiązkowe zabezpieczenia i bazy, w których przechowuje się informacje o każdym opakowaniu jednostkowym. Nowe przepisy zaczną obowiązywać w UE i EOG z dniem 9 lutego 2019 r. Od tego dnia leki na receptę wprowadzane na rynek UE będą musiały posiadać niepowtarzalny identyfikator oraz element uniemożliwiający naruszenie opakowania zgodnie z wymogami określonymi w dyrektywie w sprawie sfałszowanych produktów leczniczych oraz w rozporządzeniu delegowanym Komisji (UE) 2016/1614. System baz, który jest obecnie tworzony przez zainteresowane podmioty i który składa się z europejskiego systemu centralnego i krajowych baz danych, również będzie musiał zacząć działać najpóźniej od dnia 9 lutego 2019 r. Posiadacze pozwoleń na dopuszczenie do obrotu, producenci, hurtownicy i podmioty dostarczające produkty lecznicze ludności (w tym szpitale) będą musieli badać leki w różnych punktach łańcucha dystrybucji w celu wprowadzenia do bazy, weryfikacji autentyczności oraz wycofania z bazy w momencie ich wydania.
Apteki szpitalne i działy farmacji szpitalnej odgrywają istotną rolę w weryfikacji autentyczności produktów leczniczych dostarczanych pacjentom. Po otrzymaniu leków podmioty te muszą sprawdzić zabezpieczenia i wycofać jego niepowtarzalny identyfikator. Weryfikacja zabezpieczeń i wycofywanie niepowtarzalnych identyfikatorów będą wymagały zakupu skanerów do odczytu niepowtarzalnego identyfikatora oraz aktualizacji oprogramowania służącego łączeniu się z systemem baz. W związku z dużą ilością leków, jakimi obracają apteki szpitalne, od dnia 9 lutego 2019 r. będą one również musiały być w stanie szybko i skutecznie sprawdzać opakowania jednostkowe. Apteki szpitalne i działy farmacji szpitalnej nie będą mogły wydawać na oddziały leków z zabezpieczeniami, jeżeli nie będą w stanie zweryfikować i wycofać niepowtarzalnych identyfikatorów i muszą przewidzieć wystarczającą ilość czasu na przygotowanie się do dnia 9 lutego 2019 r.
Obowiązek podłączenia do systemu baz szpitali i funkcjonujących w nim aptek i działów farmacji szpitalnej poszczególnych krajów członkowskich Krajowej Organizacji Weryfikacji Autentyczności Leków wynika wprost z dyrektywy unijnej 2011/62/EU tzw. Dyrektywy fałszywkowej i implementującej ją do polskiego prawa ustawy z dnia 19 grudnia 2014 r. o zmianie ustawy – Prawo farmaceutyczne oraz niektórych innych ustaw (Dz. U. z dnia 8 stycznia 2015 r.) oraz aktów wykonawczych do dyrektywy.
W związku z powyższym prezes NRA Elżbieta Piotrowska – Rutkowska zwraca się z prośbą o nadanie priorytetowego znaczenia wszystkim działaniom dostosowującym Państwa szpital do wymogów dyrektywy fałszywkowej oraz zapewnienie kierownikowi apteki szpitalnej/działu farmacji szpitalnej sprzętu, oprogramowania oraz zasobów osobowych gwarantujących realizację obowiązków nałożonych rozporządzeniem delegowanym Komisji (UE) 2016/1614.
Szczegółowe informacje możecie Państwo znaleźć na stronie Krajowej Organizacji Weryfikacji Autentyczności Leków (KOWAL)
Komunikat Ministerstwa Zdrowia w sprawie Safety Features w aptekach szpitalnych z dnia 12.12.2018 roku dostępny tutaj.
Informacja o pilotażu Polskiego Systemu Weryfikacji Produktów Leczniczych
W związku z zaangażowaniem Krajowej Organizacji Weryfikacji Autentyczności Leków, której Naczelna Izba Aptekarska jest członkiem, w prace nad wdrożeniem w Polsce Dyrektywy fałszywkowej, w zakresie zabezpieczenia europejskiego łańcucha dystrybucyjnego przed wprowadzaniem sfałszowanych produktów leczniczych, w najbliższym czasie planowane jest uruchomienie pilotażu Polskiego Systemu Weryfikacji Produktów Leczniczych.
Jednym z celów Dyrektywy fałszywkowej jest stworzenie europejskiego systemu baz danych poprzez który apteki, hurtownie oraz inne podmioty dostarczające leki pacjentom będą dokonywać weryfikacji autentyczności produktów leczniczych przed ich wydaniem. W celu zwiększenia nadzoru dyrektywa wprowadziła obowiązek umieszczania na większości leków na receptę dwóch rodzajów zabezpieczeń: unikalnego identyfikatora (UI) w postaci kodu dwuwymiarowego (zdjęcie) oraz elementów wskazujących na otwarcie opakowania (ATD, anti-tempering devices).
Obowiązek weryfikacji i wycofania kodów z bazy danych został nałożony na wszystkie osoby upoważnione lub uprawnione do wydawania pacjentom produktów leczniczych objętych serializacją. Dotyczy to zarówno hurtowników, aptekarzy w aptekach ogólnodostępnych i szpitalnych, działach farmacji, jak i podmiotów wymienione w art. 23 Rozporządzenia Delegowanego 2016/161.
Mając na uwadze powyższe zachęcam Państwa do zgłoszenia aptek do udziału w pilotażu Systemu Weryfikacji Produktów Leczniczych.
Warunkiem niezbędnym do uczestnictwa w pilotażu jest posiadanie przez apteki skanerów 2D.
Zgłoszenie do uczestnictwa w pilotażu należy dokonać mailowo na adres: nia@nia.org.pl.
Dalsze szczegółowe informacje, dotyczące uczestnictwa aptek w pilotażu, będziemy przekazywać wszystkim zainteresowanym, którzy wyrażą chęć wzięcia udziału w wymienionym programie.
Szanowni Państwo, wszelkie zapytania dotyczące serializacji prosimy kierować na specjalny, dedykowany adres: serializacja@nia.org.pl
Fundacja KOWAL przekazała użytkownikom testującym dostęp do PLMVS w środowisku IQE zintegrowaną aplikację służącą weryfikacji, wycofaniu oraz cofnięciu wycofania niepowtarzalnych identyfikatorów produktów serializowanych dostępnych w środowisku IQE. Aplikacja dostępna jest pod adresem https://pilot.nmvo.pl. Całość komunikatu dotyczącego aplikacji dostępna jest tutaj.
Generowanie certyfikatu dostępu do środowiska pilotażowego IQE:
Wymagania dla czytników kodów 2D
W związku z przygotowaniami mającymi na celu wdrożenie Dyrektywy Fałszywkowej przypominamy, że do uruchomienia systemu informatycznego umożliwiającego weryfikację i wycofanie kodów z bazy niezbędny jest zakup skanerów 2D spełniających poniższe wymagania:
- Odczyt kodów 2D Data Matrix (ECC 200) oraz QR Kod.
- Odczyt kodów z wyświetlacza przenośnego urządzenia elektronicznego (tablet, smartfon, itp.)
- Możliwość wprogramowania znaku „@” jako prefix dla czytników kodów (dla aptek pracujących na systemie wspomagania obsługi aptek KS Apteka m.in. firmy KAMSOFT)
Lista oprogramowania dopuszczonego do integracji z PLMVS
Dzięki systemowi PLMVS apteki, szpitale i hurtownie będą mogły realizować swoje obowiązki związane z weryfikacją autentyczności leków lub ich dezaktywacją w systemie przed ich wydaniem pacjentowi. Aktualna lista oprogramowania dopuszczonego do integracji z PLMVS dostępna jest na stronie Krajowej Organizacji Weryfikacji Autentyczności Leków
Ważne informacje dot. uzyskania certyfikatu niezbędnego do procesu serializacji
W związku z wysyłką korespondencji elektronicznej docierającej do aptek ogólnodostępnych, aptek szpitalnych, działów farmacji, hurtowni farmaceutycznej z Fundacji KOWAL, Naczelna Izba Aptekarska przypomina, że zgodnie z załączonym komunikatem uzyskanie certyfikatu niezbędnego do weryfikacji oryginalności leków wymaga otrzymania:
- wysyłki e-mail (wiadomości e-mail)
- wysyłki pocztowej (listu poleconego tradycyjną drogą pocztową)
Uprzejmie informujemy, że korespondencja e-mail jest wysyłana w 2 etapach:
Pierwsza część pism została wysłana w dniu 21 stycznia 2019 r.
Druga część pism zostanie wysłana w dniu dzisiejszym tj. 22 stycznia 2019 r.
Uzyskanie certyfikatu wymaga wykorzystania danych (Login, hasło, numer TAN, link do portalu Arvato) z obydwu otrzymanych źródeł.
Po otrzymaniu korespondencji e-mail kolejnym krokiem koniecznym do uzyskania certyfikatu jest otrzymanie korespondencji poleconej.
UWAGA!!! W przypadku nie otrzymania od Fundacji KOWAL korespondencji e-mail 22 stycznia 2019 r. do godziny 16.00 wymagana jest aktualizacja adresu e-mail w bazie KOWAL.
W celu zapewnienia skutecznej aktualizacji adresu e-mail należy przesłać aktualny adresu e-mail wraz z numerem identyfikacyjnym – ID, widniejącym w CSIOZ na adres: plmvo.support@nmvo.pl.
Poniżej prezentujemy film przedstawiający proces pobrania certyfikatu z portalu Arvato:
Szczegółowe informacje w wersji PDF, jak uzyskać certyfikat, przygotowane przez Fundację Kowal można pobrać tutaj
Komunikat prezesa Fundacji KOWAL Michała Kaczmarskiego ws. produktów, które zostały wprowadzone do obrotu przed 9 lutego 2019 roku
Fundacja KOWAL informuje o statusie certyfikacji
Krajowa Organizacja Weryfikacji Autentyczności Leków (Fundacja KOWAL) sfinalizowała dwuetapową certyfikację – wysyłając drogą pocztową oraz e-mailową dane dostępowe do pobrania certyfikatów do wszystkich Użytkowników Końcowych, figurujących w bazie CSIOZ.
Aktualnie zespół Fundacji pracuje nad bieżącym uzupełnianiem nadesłanych przez Użytkowników Końcowych adresów e-mail, których brakowało w Rejestrach Medycznych CSIOZ lub były podane w rejestrach błędnie. Zespół weryfikuje również otrzymane zwroty, ponownie przesyłając korespondencję dostępową.
Komunikat w sprawie produktów wyłączonych z serializacji i wymienionych w Załączniku nr i do Rozporządzenia Delegowanego Komisji (UE) 2016/161, które posiadają kody 2D Data Matrix
Krajowa Organizacja Weryfikacji Autentyczności Leków przekazała informację do Naczelnej Izby Aptekarskiej o wzroście zapytań farmaceutów, dotyczących produktów wyłączonych z serializacji i wymienionych w Załączniku Nr I do Rozporządzenia Delegowanego Komisji (UE) 2016/161, które posiadają kody 2D Data Matrix.
Uprzejmie wyjaśniamy, że są to głównie produkty o kodzie ATC nie podlegające serializacji.
Z informacji uzyskanych od Krajowej Organizacji Weryfikacji Autentyczności Leków wynika, że produkty te nie podlegają serializacji, a co za tym idzie nie znajdują się w bazie PLMVS. W związku z tym każdorazowe skanowanie tych produktów wygeneruje komunikat o treści „Nieznany kod produktu.”
Do pobrania: Załącznik nr 1 do Rozporządzenia Delegowanego Komisji (UE) 2016161
Wyjaśnienia i zestawiania dotyczące możliwych informacji przekazywanych przez PLMVO do systemów użytkowanych przez apteki ogólnodostępne, szpitalne oraz hurtownie farmaceutyczne
Uprzejmie informujemy, że na stronie internetowej Fundacji KOWAL – https://www.nmvo.pl/pl/aktualnosci/jakie-informacje-po-zeskanowaniu-leku-wysyla-plmvs/ zostały zamieszczone wyjaśnienia i zestawiania dotyczące możliwych informacji przekazywanych przez PLMVO do systemów użytkowanych przez apteki ogólnodostępne, szpitalne oraz hurtownie farmaceutyczne.
W zależności od etapu skanowania oraz zawartości przekazywane informacje zostały podzielone na:
- Status – informuje o statusie paczki w systemie (Załączona tabela zawiera informacje o sposobie postępowania). Wyświetlany jest na etapie weryfikacji paczki.
- Komunikat – dotyczy kwestii technicznego administrowania kontem w systemie PLMVS (Załączona tabela zawiera informacje o sposobie postępowania).
- Alert – informuje o niezgodności zeskanowanych danych z informacjami zawartymi w PLMVS (Sposób postępowania w przypadku wystąpienia alertu znajduje się w przewodniku GIF – https://www.gov.pl/web/gif/elektroniczne-raportowanie-do-plmvo).
Jednocześnie informujemy, że Naczelna Izba Aptekarska prowadzi prace mające na celu zwiększenie czytelności informacji przekazywanych przez systemy informatyczne użytkowane przez farmaceutów.
Do pobrania:
Przewodnik zarządzania alertami w PLMVS
Kliknij w obraz, żeby powiększyć:
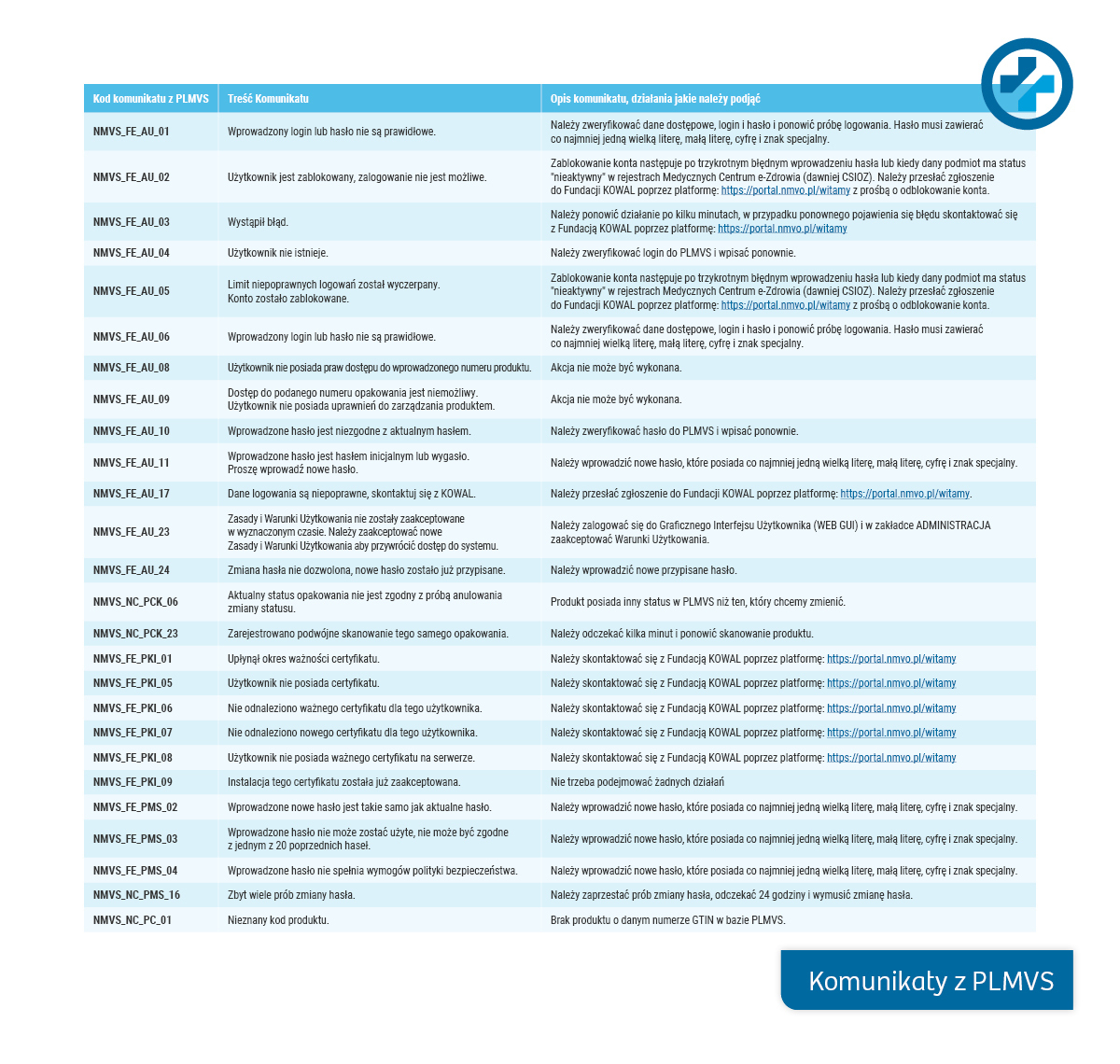
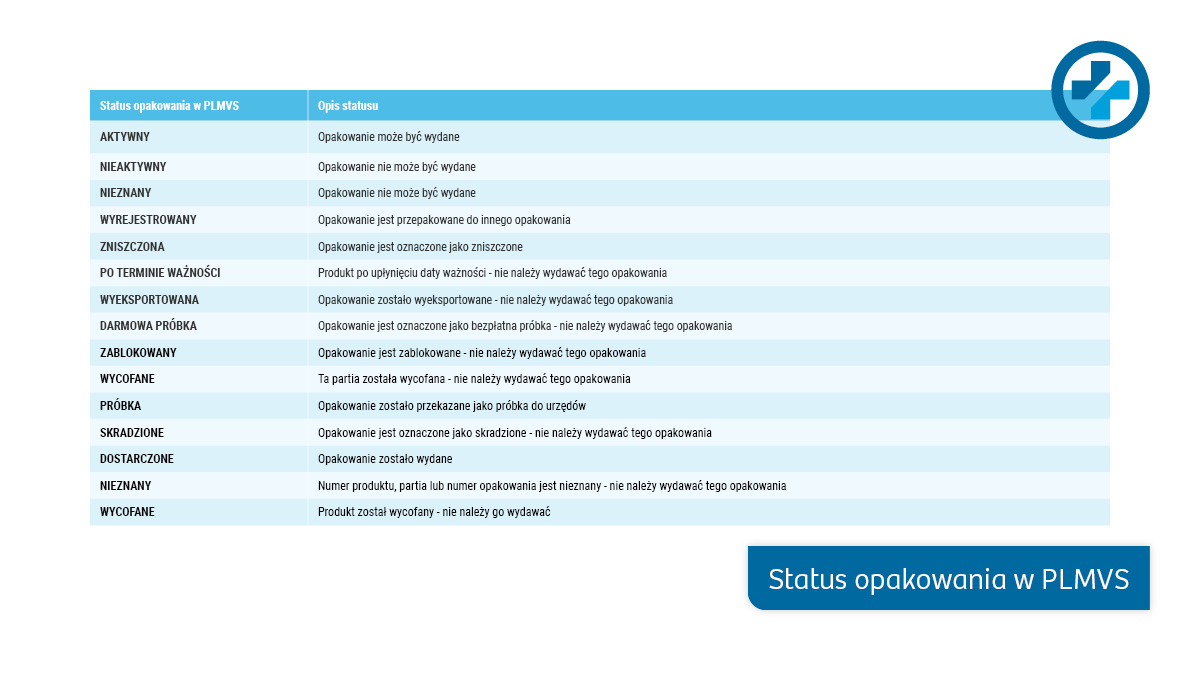

PLMVS-Najczęściej pojawiające się pytania
W odpowiedzi na najczęściej pojawiające się kwestie dotyczące systemu PLMVS Zespół Fundacji KOWAL przygotował zbiór odpowiedzi, które pomogą wyjaśnić poszczególne zagadnienia.
Przejdź do poradnika.













